
Рефераты по биологии
Рефераты по экономике
Рефераты по москвоведению
Рефераты по экологии
Краткое содержание произведений
Рефераты по физкультуре и спорту
Топики по английскому языку
Рефераты по математике
Рефераты по музыке
Остальные рефераты
Рефераты по авиации и космонавтике
Рефераты по административному праву
Рефераты по безопасности жизнедеятельности
Рефераты по арбитражному процессу
Рефераты по архитектуре
Рефераты по астрономии
Рефераты по банковскому делу
Рефераты по биржевому делу
Рефераты по ботанике и сельскому хозяйству
Рефераты по бухгалтерскому учету и аудиту
Рефераты по валютным отношениям
Рефераты по ветеринарии
Рефераты для военной кафедры
Рефераты по географии
Рефераты по геодезии
Рефераты по геологии
Реферат: Алкилирование енаминов, бета-дикетонов и енаминокетонов
Реферат: Алкилирование енаминов, бета-дикетонов и енаминокетонов
Новосибирский Государственный Университет
Кафедра Органической Химии
Курсовая работа по органической химии
студента II курса ФЕН Купрова И.С.
Тема: Алкилирование енаминов, β-дикетонов и β-енаминокетонов.
Научный руководитель:
д.х.н. проф. Ткачев А.В.
Новосибирск, 2000.
"The true worth of an experimenter
consists in his pursuing not only
what he seeks in his experiment,
but also what he did not seek."
Claude Bernard
Введение.
Классический подход к формированию связи углерод-углерод – реакция “нуклеофил плюс электрофил” – в настоящее время принимает самые разнообразные формы и является одним из основных инструментов синтетической органической химии. В качестве нуклеофилов в этих реакциях могут выступать разнообразные соединения, несущие неподеленную пару электронов, отрицательный заряд, либо активированную ароматическую систему. Во многих случаях нуклеофильность тесно соседствует с основностью – сродством к протону, которое может осложнять проведение нуклеофильных реакций за счет побочных процессов элиминирования.
В плане легкости образования карбаниона и одновременно высокого отношения нуклеофильность/основность из синтетически значимых нуклеофильных групп наибольшего внимания заслуживает атом углерода, находящийся на конце сопряженной системы, включающей в себя гетероатом (как правило, N либо O). Нуклеофильные свойства в этом случае обусловлены наличием неподеленной пары электронов и/или делокализованным отрицательным зарядом:
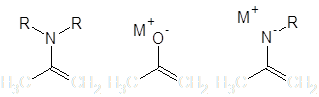
Соединения, содержащие подобные группировки широко используются как нуклеофилы в реакциях алкилирования. В этих структурах, однако, присутствуют два сравнимых по нуклеофильности центра – аллильный углерод и гетероатом, и большинство реакций в той или иной мере проходит по обоим этим положениям.
Введение в состав молекулы одновременно двух групп, стабилизирующих отрицательный заряд, существенно изменяет ее поведение в нуклеофильных реакциях, в частности, после подбора температуры и кислотности среды, оказывается возможным проведение селективного С-алкилирования. Интересным и синтетически значимым примером подобных соединений являются моноимины β-дикетонов, обычно существующие в термодинамически более стабильной енаминной форме:
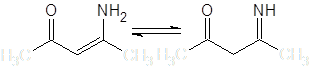
В настоящей работе сделан обзор литературных данных по реакциям С-алкилирования упомянутых групп соединений и исследована реакция бензилирования (3-амино-6,6-диметилбицикло[3.1.0]гекс-2-ен-2-ил)метилкетона – енаминокетона, получаемого из природных терпеноидов.
Енамины, кетоны, β-дикетоны.
Синтезу и общим свойствам енаминов посвящена монография Дайка [1]. Как правило, енамины получаются взаимодействием карбонильных соединений с вторичными аминами (при получении с целью последующего алкилирования – обычно циклическими). В водных средах они гидролизуются, регенерируя исходные карбонильные соединения [2]:
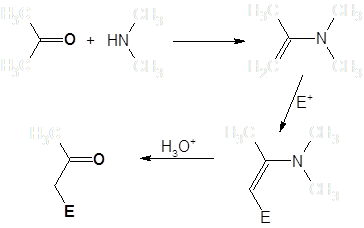
Скорости образования енаминов для различных вторичных аминов в большинстве случаев образуют следующий ряд:
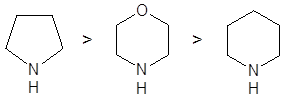
Условия протекания реакций алкилирования качественно одинаковы для всех рассматриваемых групп – щелочная среда или условия способствующие депротонированию α-углерода. Амминный атом водорода енаминов, также как Hα-С, в некоторой степени обладает кислотными свойствами, и в сильнощелочной среде реакция по гетероатому может конкурировать с основной. Особенно выделяются вторичные енамины, которые после N-депротонирования выступают как весьма активные N-нуклеофилы:
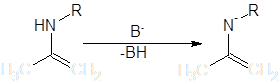
При их обработке электрофилом образуется от 10 до 70% N-замещенного продукта и многие авторы рассматривают C-алкилирование этих соединений лишь как побочную реакцию [3]. В целом, “семейство” побочных реакций для процессов нуклеофильного замещения довольно обширно, основные могут быть сведены в приведенную ниже схему (на примере енолятов) [4]:
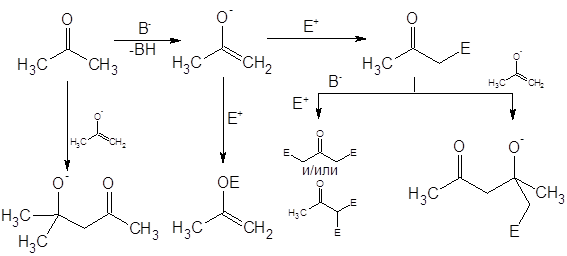
Авторы [4] приводят следующие условия как необходимые для протекания реакции алкилирования по требуемому направлению:
1. Скорость образования нуклеофильной частицы должна быть выше скорости ее конденсации с исходным веществом.
2. Селективное протекание реакции по атому углерода.
3. Скорость реакции взаимодействия нуклеофил-электрофил должна быть выше скорости реакции переноса протона.
4. Реакция нуклеофил-электрофил должна протекать быстрее, чем взаимодействие нуклеофила с продуктом реакции.
Из трех упомянутых в заголовке типов алкилируемых соединений безусловно лучшим или более удобным нельзя назвать ни один. Преимущества каждого описаны ниже:
Енамин |
Не требует высокоосновных/кислотных сред для образования или реакции. |
| Енолят (лития) | Легко и быстро генерируется, реагирует с предсказуемой региоселективностью. |
| Анион иминия | Исключительно низка скорость протонного переноса, предсказуемая региоселективность. |
Две основные проблемы, возникающие в реакциях алкилирования енаминов – полизамещение и реакции по атому азота, наиболее наглядно проявляют себя в случае алкилирования алкилгалогенидами. Это проиллюстрировано результатами настоящей работы и данными авторов [4] (метилирование), опубликовавших системные исследования по этой тематике:
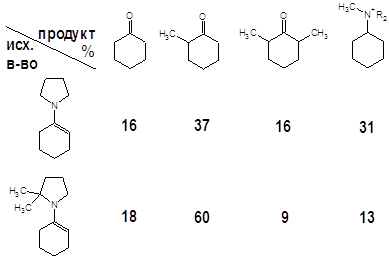
Вторая строчка таблицы несколько “лучше” – за счет наличия двух метильных групп реакция протекает более или менее однозначно. Стерические затруднения, характерные для более сложных кетонов, снижают долю полиалкилированых продуктов, хотя и увеличивают время реакции [4]. Те же пространственные факторы во многих кетонах ограничивают список потенциальных алкилирующих агентов следующими:
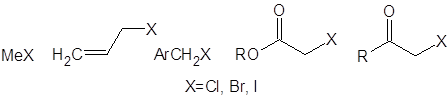
Даже с этими соединениями алкилирование кетонов через еноляты лития обеспечит высокий выход лишь для кетонов, устойчивых в сильноосновной среде (необходимой для генерирования енолятов). В случае β-дикетонов для депротонирования требуется менее основная среда. В работе [5] исследовано влияние пространственного строения бокового алкильного заместителя на скорость и региоселективность реакций алкилирования β-дикетонов:
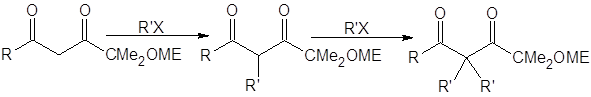
В противоположность алкилированию галогенидами, присоединение по Михаэлю активированных алкенов проходит с высокой специфичностью по атому углерода. Реакция по гетероатому в данном случае оказывается обратимой и сдвиг к термодинамически более стабильному продукту С-алкилирования приводит к хорошим выходам соответствующих алкилпроизводных:
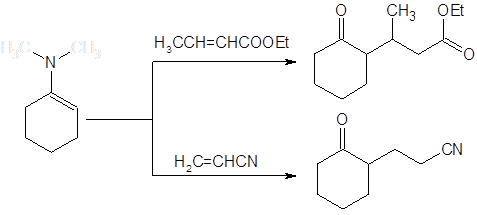
В отличие от кето-енольной таутомерии, факторы, контролирующие енамин-иминные превращения, изучены мало. Известно, что стабильность енаминных форм выше для третичных и ниже для первичных ненасыщенных аминов, но даже первичные и вторичные енамины могут быть стабилизированы введением в β-положение соответствующего заместителя (в работе [6] – COOH). Показано также, что полярные растворители сдвигают равновесие в сторону образования енаминов. Данные теоретических расчетов, в то же время, дают противоположный результат [6]. Для некоторых катион-радикалов енаминов и иминов и соответствующих нейтральных молекул измерены теплоты образования [7].
Енамин-иминная таутомерия может обуславливать быструю рацемизацию некоторых веществ в протонных растворителях, препятствуя тем самым разделению энантиомеров соединений, содержащих в углеродном скелете группировку
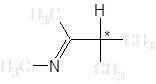
Авторы [8] наблюдали рацемизацию (S)-тетрагидрозолина, обусловленную таутомерными переходами:
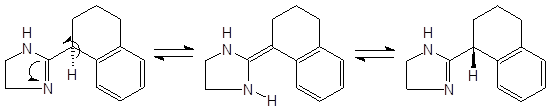
Исследование кинетики реакции рацемизации показало, что содержащие протон асимметрические центры по соседству с иминной группой “долго не живут”.
Реакции ароматических енаминов могут катализироваться тетрагалогенидами Ti, Zr, Hf [9]. В качестве любопытного примера реакции алкилирования енамина можно привести осуществленную совсем недавно межлигандную конденсацию [10]:
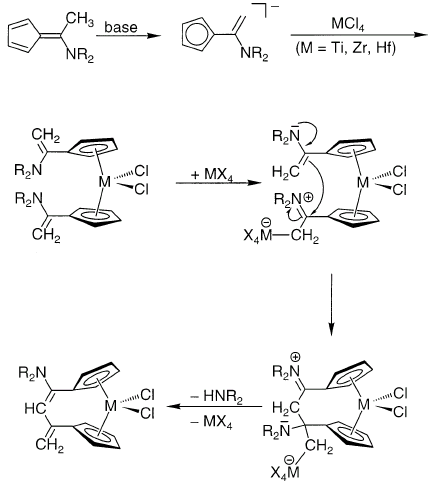
β-енаминокетоны.
Один из наиболее удобных синтетических методов получения сложных енаминонов, в том числе оптически активных – синтез на основе природных терпеноидов – лимонена, 3-карена и δ-кадинола. Эти соединения, интересные сами по себе (отмечена их биологическая активность), являются ключевыми промежуточными продуктами в асимметрическом синтезе [11]. Енаминокетоны, полученные из этих терпенов, используются как хиральные основания для разделения энантиомеров оптически активных кислот [12].
Енаминная система в щелочных средах может быть депротонирована и продукт введен в реакцию с алкилирующими агентами.
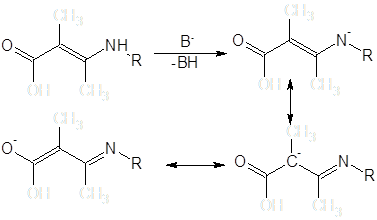
До последнего времени считалось, что преобладающими продуктами реакций алкилирования енаминокетонов являются N-замещенные производные [13]. Данные последующих исследований показывают, что алкилирование некоторых β-енаминокетонных систем в условиях межфазного переноса может быть селективно проведено и по атому углерода [14]:
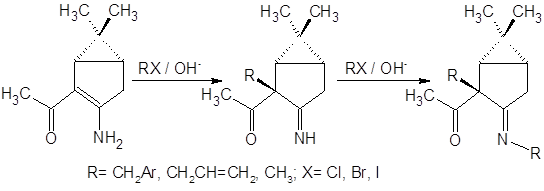
Приведенная последовательность превращений характерна, однако, только для алкилирования стерически нагруженными алкилгалогенидами. При использовании в качестве галогенида йодистого метила образуются все продукты вплоть до пентазамещенного. Исходя из данных настоящей работы, можно отметить, что направление алкилирования приведенных на рисунке соединений существенно зависит также от температуры реакции: при 35°С преобладает продукт С-алкилирования, при более низких температурах из смеси удается выделить N-алкилзамещенный продукт, доля С-замещения при этом невелика.
В работе [13] исследовано алкилирование 3-амино-5,5-диалкилциклогекс-2-ен-1-она для различных алкильных заместителей и проведен анализ факторов, необходимых для селективного направления алкилирования по тем или иным положениям изученного енаминона:
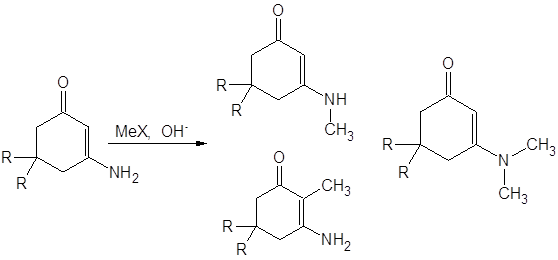 Зависимость хода реакции
алкилирования аналогичного [13] циклического енаминокетона от природы боковых
радикалов изучена авторами [3]:
Зависимость хода реакции
алкилирования аналогичного [13] циклического енаминокетона от природы боковых
радикалов изучена авторами [3]:
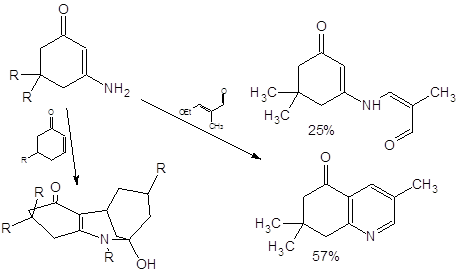
Среди известных реакций енаминонов внимания также заслуживает описанная в [15] реакция фотоарилирования:
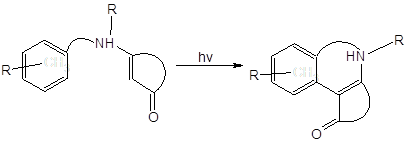
Авторы утверждают, что облучение енаминонов светом с длиной волны < 300 нм “can result in the formation of a variety of products… …photoreductions predominate”.
Из приведенных литературных данных можно сделать выводы об условиях, необходимых для получения высоких выходов С-алкилированных производных енаминов, b-дикетонов и енаминонов:
1. Необходим подбор основности среды. В низкоосновных средах мала концентрация активного аниона и реакция протекает медленно, в слишком высокоосновной среде происходит депротонирование атома азота и преобладающим становится продукт N-алкилирования.
2. Упомянутые выше требования к кинетике процесса алкилирования должны быть удовлетворены в максимальной степени.
3. С-алкилированный продукт, получающийся при повышенных температурах, является следствием термодинамического контроля реакции реакции алкилирования, при снижении температуры реакции возрастает доля продуктов кинетического контроля -- N- и O-алкилированных продуктов.
4. Асимметрическая индукция от имеющихся структурных фрагментов может обеспечивать отмеченное многими авторами стереоселективное протекание реакции алкилирования [2, 3, 5, 11, 12, 14].
В целом можно отметить, что несмотря на широкую известность описанных соединений, реакции алкилирования с их участием изучены пока недостаточно.
![]()
Экспериментальная часть.
Синтез (3-амино-6,6-диметилбицикло[3.1.0]гекс-2-ен-2-ил)метилкетона II:
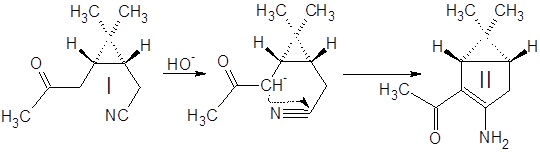
К 350 мл спиртового раствора KOH при перемешивании в течение 10 мин. добавили 100 г кетонитрила I. Смесь нагрели и кипятили с обратным холодильником 15 мин., охладили и разбавили водой в три раза. Провели экстракцию смеси метилтретбутиловым эфиром (300, 300, 150 мл), эфирную фазу экстрагировали 1М HCl (900, 500, 300 мл). Полученный водный раствор нейтрализовали 30% аммиаком и экстрагировали tBuOMe (200, 200, 100 мл). Эфирный раствор высушили MgSO4 безв и отогнали растворитель. Выход 79%.
Синтез (3-амино-2-бензил-6,6-диметилбицикло[3.1.0]гекс-2-ил)метилкетона III:
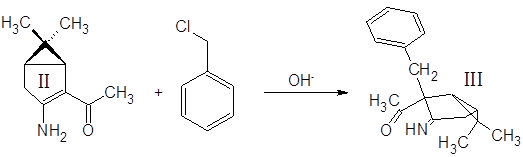
5 г енаминокетона II растворили в 30 мл бензола, в раствор добавили 15 мл 40% водного раствора NaOH и 0.5 г Bu4N+NO-3, перемешивали несколько минут и постепенно (3 мин.) добавили 10 мл бензилхлорида. Смесь интенсивно перемешивали 1.5 часа при 35-40˚С. Водную фазу отбросили, органическую экстрагировали 1М H2SO4 (15, 15, 15 мл). Экстракт нейтрализовали избытком 30% аммиака и экстрагировали метилтретбутиловым эфиром. Эфирный раствор высушили MgSO4 безв и отогнали растворитель. Выход 44%.
Синтез 1-(3-амино-6,6-диметилбицикло[3.1.0]гекс-2-ен-2-ил)этанола:
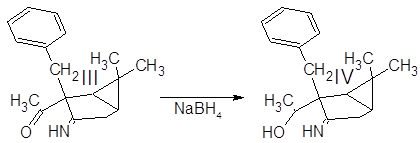
4 г алкилированого енаминокетона III растворили в 40 мл этанола. В раствор всыпали 0.6 г NaBH4 и перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь разбавили водой и экстрагировали эфиром. Эфирную фазу промыли водой для удаления спирта и вылили в водный раствор 4 г Cu(OAc)2·2H2O. Голубые кристаллы отфильтровали и высушили на воздухе.
Продукты исследовали методами хроматомасс-спектрометрии, ИК и ЯМР 13C и 1H. ЯМР спектры регистрировали на приборе Bruker DPX-500 (НИОХ СО РАН) в смеси CCl4/CDCl3; химсдвиги отсчитывали: в протонных спектрах – от сигнала остаточных протонов CDCl3 (7.250 м.д.), в спектрах 13С – от сигнала атома углерода CCl4 (96.10 м.д.). ИК спектры записывали на однолучевом спектрометре Bruker Vector 22 (256 усреднений с вычетом фона). Хроматомасс-спектрометрический анализ выполнен сотрудниками НИОХ.
Бензилхлорид и все растворители использовали свежеперегнанными. Точность отсчета температуры ±2°С, времени – ±2 мин. Тонкослойная хроматография выполнена на пластинках “Silufol”® (SiO2 на алюминиевой фольге).
Результаты и их обсуждение.
Из описанных выше реакций С-алкилирования наилучшим образом изучены реакции алкилирования b-дикетонов и енаминов. По енаминокетонам, несмотря на их широкое применение в синтезе, данных значительно меньше. Практическая потребность в проведении алкилирования и обнаруженная неоднозначность протекания этой реакции потребовали ее более детального изучения на конкретных соединениях. В качестве алкилирующего реагента был избран бензилхлорид, в условиях реакции не дающий продуктов полиалкилирования (метилирование в тех же условиях может быть четырех-пятикратным [14]).
Схема проведенных превращений такова:
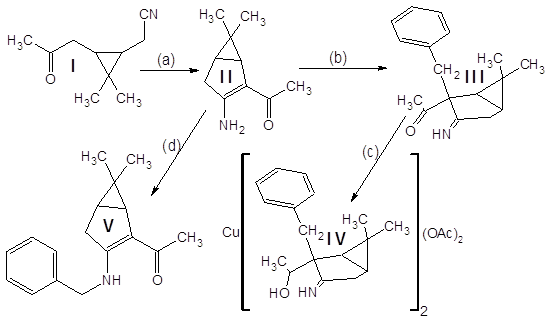
Кетонитрил I (исходное соединение, чистота ~90% (ГЖХ), предоставлен ЛТС НИОХ СО РАН) является производным природного терпена – 3-карена, выделяемого из соснового скипидара:
Результирующее соединение IV используется далее в синтезе оптически активных комплексных соединений, используемых в асимметрическом катализе.
Кетонитрил I представляет собой вязкую темную жидкость, растворимую в органических растворителях и нерастворимую в воде. Реакция (а) проходит гладко и с высоким выходом (~80%) дает продукт конденсации – (3-амино-6,6-диметилбицикло[3.1.0]гекс-2-ен-2-ил)метилкетон II.
Отдельного рассмотрения заслуживает процесс бензилирования соединения II. Эта реакция при комнатной температуре протекает крайне неоднозначно, с выходом целевого продукта менее 10% и образованием трудноразделимой смеси изомерных продуктов и продуктов полиалкилирования. Схема превращений и идентифицированные продукты представлены на следующем рисунке:
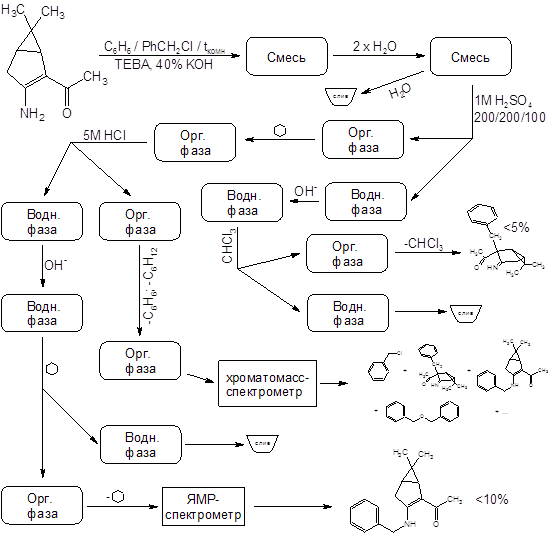
Показанные на схеме процессы обладают повторяемостью – по данным тонкослойной хроматографии в продуктах нескольких проведенных реакций набор компонентов один и тот же. Выходы выделенных веществ тоже примерно одинаковы. Спектры ядерного магнитного резонанса N-алкилированого продукта имеются в Приложениях. Его выход составляет 10%, но (!) – зимой. В тех же реакциях, осуществленных летом, продукт N-алкилирования зафиксирован не был.
Предположение о связи направления алкилирования с температурой реакционной смеси (а в методике указана комнатная температура) подтвердилось – проведение реакции при 35ºС дало в качестве преобладающего продукта (с выходом ~60%) целевой С-замещенный енаминокетон, алкилирование прошло так, как описано в “летней” методике. Попытки дальнейшего повышения температуры пока не предпринимались.
Полученный (3-амино-2-бензил-6,6-диметилбицикло[3.1.0]гекс-2-ил)метилкетон III был введен в реакцию с избытком боргидрида натрия для восстановлния карбонильной группы в спиртовую.
Точный состав и строение комплекса IV неизвестны – парамагнитный ион меди II препятствует получению спектров ЯМР, по данным ИК-спектра что-либо определенное сказать затруднительно. Сводка полученных характеристик соединений дана ниже:

Результаты проделанной работы можно суммировать следующим образом:
- синтезированы и охарактеризованы спектрально и хроматографически некоторые производные природного терпена 3-карена.
- обнаружена значительная зависимость хода реакции бензилирования (3-амино-6,6-диметилбицикло[3.1.0]гекс-2-ен-2-ил)метилкетона от температуры реакционной смеси. Установлено, что оптимальный выход С-бензилированного производного достигается при t≥35˚C.
- установлен тот факт, что “комнатная” температура – весьма ненадежная характеристика методики синтеза.
![]()
Литература.
1. S. Dyke // Chemistry of enamines. // Cambridge University Press, Cambridge, 1973.
2. A. Cook (ed) // Enamines: synthesis, structure and reactions. // Marcell Decker, New York, 1969.
3. I. Chaaban, J. Greenhill, M. Ramli // Reactions between enaminones and enones. Part 2. C versus N-alkylation with cyclohex-2-enone. Structure confirmation by reduction of a dienaminone derivative of a dehydrated dimedone dimer. // J. Chem. Soc. Perkin Trans., 1981, 1, 3120-3124.
4. J. Whitesell, M. Whitesell // Alkylation of ketones and aldehydes via their nitrogen derivatives. // Synthesis, 1983, 7, 517-536.
5. A. Zanina, S. Shergina, I. Sokolov, M. Shvartsberg // Alkylation of sterically hindered 1,3-diketones under phase-transfer conditions. // Russ. Chem. Bl., 1996, 45, 2389-2392 // Izv. Akad. Nauk Ser. Khim. 1996, 10, 2518-2521.
6. S. Lu, A. Lewin // Enamine-imine tautomerism in α,β-unsaturated α-amino acids. // Tetrahedron, 1998, 54, 15097-15014.
7. J. Henriksen, S. Hammerum // Heats of formation of imine and enamine radical cations and the corresponding neutral molecules // Int. J. Mass Spect, 1998 179/180, 301-308.
8. S. Caccamese, G Principato // Resolution of the enantiomers of tetrahydrozoline by chiral HPLC. The racemization of the enantiomers via an imine-enamine tautomerism. // Tetrahedron: Asymmetry, 1998, 9, 2939-2945.
9. M. Shimizu, A. Morita, T. Kaga // Double nucleophilic addition to α,β-unsaturated aldimines induced by titanium tetrahalides. // Tetrahedron Lett., 1999, 40, 8401-8405.
10. S. Knüppel, R. Frölich, G. Erker // Formation of functionalized [3]ferrocenophane derivatives by an enamine condensation reaction.// J. Organomet. Chem., 2000, 595, 308-312.
11. A. Tkachev, A. Rukavishnikov // Enaminones of the 2-acetylcyclopent-1-en-1-ylamine type derived from the terpenic compounds limonene, 3-carene and δ-cadinol. // Mendeleev Commun., 1992, 1, 161-162.
12. S. Popov, A. Tkachev // New chiral agents for resolution of racemic cis-permethric and cis-Z-cyhalothric acids. // Tetrahedron: Asymmetry, 1995, 6, 4, 1013-1018.
13. J. Greenhill, A. Moten // N-Alkylation of enaminones. // Tetrahedron, 1983, 39, 3405-3408.
14. A. Tkachev, S. Popov // Alkylation of enaminoketone with a modified carane skeleton. Formation of stable β-diketone monoimines. // Russ. J. Org. Chem., 1997, 33, 5, 601-606.
15. T. Tiner-Harding, P. Mariano // Intramolecular photoarylations of N-(haloaryl)ethyl-β-enaminones. // J. Org. Chem., 1982, 47, 482-485.
16. P. Bugler, I. Cottrell, C. Cowden, A. Davies, U. Dolling // An investigation into the alkylation of 1,2,4-triazole // Tetrahedron Lett., 2000, 41, 1297-1301.
Приложение 1.
Данные и аннотации некоторых статей (Belstein Abstracts).
Alkylation of Enaminoketone with a Modified Carane Skeleton. Formation of Stable b-Diketone Monoimines
A. V. Tkachev; S. A. Popov
Source details: Russ.J.Org.Chem. 1997, 33 : 5 601-606.
Document type: Journal
CODEN: RJOCEQ
Language: EN
CNR: 6090629
Original Source: Zh.Org.Khim. 1997, 33 : 5 660-665.
CODEN: ZORKAE
Language: RU
Abstract
Alkylation of a bicyclic enaminoketone, 1-((1R,5R)-3-amino-6,6-dimethylbicyclo[3.1.0]hex-2-en-2-yl))ethanone, with highly reactive alkyl halides (methyl iodide, benzyl halides, and allyl halides) in a two-phase system benzene-40% aqueous NaOH in the presence of benzyltriethylammonium chloride (BTEA) as phase-transfer catalyst results in formation of stable b-diketone monoimines, derivatives of the series of 1-((1R,5R)-2-alkyl-3-imino-6,6-dimethylbicyclo[3.1.0]hex-2-yl)ethanone, in 53-81% yields.
Alkylation of sterically hindered 1,3-diketones under phase-transfer conditions
A. S. Zanina; S. I. Shergina; I. E. Sokolov; M. S. Shvartsberg
Source details: Russ.Chem.Bl. 1996, 45 : 10 2389-2392.
Document type: Journal
CODEN: RCBUEY
Language: EN
CNR: 6056087
Original Source: Izv.Akad.Nauk Ser.Khim. 1996, 10 2518-2521.
CODEN: IASKEA
Language: RU
Abstract
Sterically hindered 1,3-diketones react selectively with propargyl and allyl bromides under conditions of phase transfer catalysis to give C-alkylated products, whereas reactions with butyl and benzyl chlorides yield mixtures of C- and O-isomers.An increase in the size of the substituents present in the initial 1,3-diketone hampers introduction of the second propargyl group.The propargyl-substituted 1,3-diketones undergo cyclization under the alkylation conditions to give substituted furans.
Sequence of alkylation of cyclohexane-1,3-dione. Alternative synthesis of (+/-)-angustione
A. A. Zenyuk; L. G. Lis; L. I. Ukhova
Source details: Chem.Nat.Compd.(Engl.Transl.) 1991, 27 : 4 400-403.
Document type: Journal
CODEN: CHNCA8
Language: EN
CNR: 5645800
Original Source: Khim.Prir.Soedin. 1991, 4 460-463.
CODEN: KPSUAR
Language: RU
Abstract
A method is proposed for introducing one, two, or three alkyl substituents into positions 4 and 6 of the cyclohexane-1,3-dione molecule by successive alkylation under the action of strong bases. (+/-)-Angustione (a natural -diketone) has been synthesized.
A Tandem Amino-Cope Rearrangement/Enamine Alkylation Reaction
Steven M. Allin; Martin A. C. Button; Stephen J. Shuttleworth
Source details: Syn.Lett. 1997, 6 725-727.
Document type: Journal
CODEN: SYNLES
Language: EN
CNR: 6086563
Abstract
Thermally induced (3,3)-sigmatropic rearrangement of 3-amino-1,5-diene substrates occurs to give the corresponding enamine products in high yield and with excellent trans:cis enamine selectivity.The enamine produced during the amino-Cope rearrangement has been directly derivatized, representing the first report of a tandem amino-Cope rearrangement/enamine alkylation reaction.The potential of this novel synthetic strategy is outlined.
N-alkylation of enaminones
John V. Greenhill; Ashraf M. Moten
Source details: Tetrahedron 1983, 39 : 20 3405-3408.
Document type: Journal
CODEN: TETRAB
Language: EN
CNR: 5609239
Abstract
The base catalyzed N-alkylation of a series of primary and secondary enaminones has been examined in detail.The enaminone anion was found to be a weak nucleophile.Best results were obtained in tetrahydrofuran or dioxane with sodium hydride and an alkyl iodide.
Alkylation of Ketones and Aldehydes via their Nitrogen Derivatives
James K. Whitesell; Marilyn A. Whitesell
Source details: Synthesis 1983, 7 517-536.
Document type: Journal
CODEN: SYNTBF
Language: EN
CNR: 5572282
Abstract
In this review, methods for the alkylation of the aldehydes and ketones via formation of nitrogen derivatives such as enamines, metallated imines (imine anions), metallated N,N-dialkylhydrazones (N,N-dialkylhydrazone anions), dimetallated oximes (oxime dianions), and metallated O-methyloximes (O-methyloxime anions) are described.Scope, limitations, and advantages or disadvantages of the methods over comparative reactions of enolates are also mentioned.
Intramolecular Photoarylations of N-(Haloaryl)ethyl β-Enaminones
Tammy Tiner-Harding; Patrick S. Mariano
Source details: J.Org.Chem. 1982, 47 : 3 482-485.
Document type: Journal
CODEN: JOCEAH
Language: EN
CNR: 5570323
Abstract
The photochemistry of several N-(haloaryl)ethyl β-enaminones was investigated in order to develop methods for preparation of tricyclic enaminone systems.The efficiencies of intramolecular photoarylations of the haloaryl systems were found to be dependent upon the wavelength of irradiation.Accordingly, irradiations of the haloaryl β-enaminones 9a,c,d,f with Pyrex-filtered light leads to formation of the reduced N-cyclized and C-cyclized products 9b or 9e, 8a or 8b, and 10a or 10b, respectively. The major products in these processes are the reduced materials.In contrast, irradiations of the bromoaryl enaminones 9c or 9f with Vycor-filtered light results in high yielding conversions to the C-cyclized tricyclic enaminones 10a and 10b in synthetically useful yields ranging from 50% to 85%.A discussion of reasons for these wavelength dependencies is given in terms of excited-state discrimination in these bichromophoric systems.Possible reaction mechanisms are considered.The origin of another major product, 11, generated by irradiation of 9f with Vycor-filtered light, is also discussed.
Regioselective control of N-aryl enaminone alkylation
Denise Dugat; Daniel Gardette; Jean-Claude Gramain; Bertrand Perrin
Source details: Bull.Soc.Chim.Fr. 1994, 131 : 1 66-70.
Document type: Journal
CODEN: BSCFAS
Language: EN
CNR: 5851695
Abstract
The regioselectivity of the C-alkylation of unsubstituted N-aryl enaminones prepared from cyclohexane-1,3-dione may be controlled by the nature of the base used.Our results indicate that monoalkylation is completely regioselective; lithium diisopropylamide leads to α’-alkylated compounds while lithium bis(trimethylsilyl)amide affords γ-alkylated products.In contrast, alkylation of α’- or γ-substituted compounds always occurs in the α’-position regardless of the base.
Regiospecificity and Regioselectivity of the Alkylation, Acylation, Sulfenylation and Sulfonylation of Pyrrolidine Enaminones
Thomas Burgemeister; Gerd Dannhardt; Ernst Eibler; Brigitte Paulus; Klaus Ziereis
Source details: Arch.Pharm.(Weinheim Ger.) 1988, 321 : 345-348.
Document type: Journal
CODEN: ARPMAS
Language: GE
CNR: 5807249
Abstract
Different methods to modify a pyrrolidine enaminone regiospecifically or regioselectively at the N- and C-atom, respectively, are described.All compounds have been isolated and characterized, in case of the alkylation reactions the ratio of regioisomers is determined by HPLC.NOE experiments are performed in order to prove the configuration of some adducts.
Reactivity of N-substituted enaminones with unsaturated carbonyl compounds
Esther Caballero; Blanca Madrigal; Manuel Medarde; Pilar Puebla; Zoila Honores; et al.
Source details: Ach.Mod.Chem. 1998, 135 : 4 457-474.
Document type: Journal
CODEN: ACMCEI
Language: EN
CNR: 6101721
Abstract
The reactivity of enaminones of 2-aminoethanol and 2-aminoethanothiol towards polyelectrophilic reagents has been studied.A brief overview of our previous results with this enaminones, which allowed us to describe new synthesis of fused heterocycles, is included. In the present work, only the formation of C-C bond is a common feature of the reaction, to produce derivatives or simple heterocycles.Although no fused heterocycles were obtained, the reaction products have complex functionalization which will allow further cyclization to such a class of polyfused systems.
Reactions between Enaminones and Enones. Part 2. C versus N-Alkylation with Cyclohex-2-enone. Structure Confirmation by Reduction of a Dienaminone Derivative of Dehydrated Dimedone Dimer
Ibrahim Chaaban; John V. Greenhill; Mohamed Ramli
Source details: J.Chem.Soc.Perkin Trans.1 1981, 3120-3124.
Document type: Journal
CODEN: JCPRB4
Language: EN
CNR: 5625982
Abstract
Primary and secondary enaminones derived from cyclohexane-1,3-dione, dimedone, and acetylacetone react with cyclohexenone to give exclusively C-alkylated derivatives.In every case the products form carbinolamines which exist as 1-hydroxy-2-azacyclo[3.3.1]nonenes.This was confirmed in some examples by formation of an extra ring between nitrogen and oxygen.A series of dienaminones were prepared from 2-(5,5-dimethyl-3-oxocyclohex-1-enyl)-5,5-dimethylcyclohexane-1,3-dione and one of these was reduced to give an azanonene identical with that from C-alkylation.
